Architecture Design Process
Basic structure
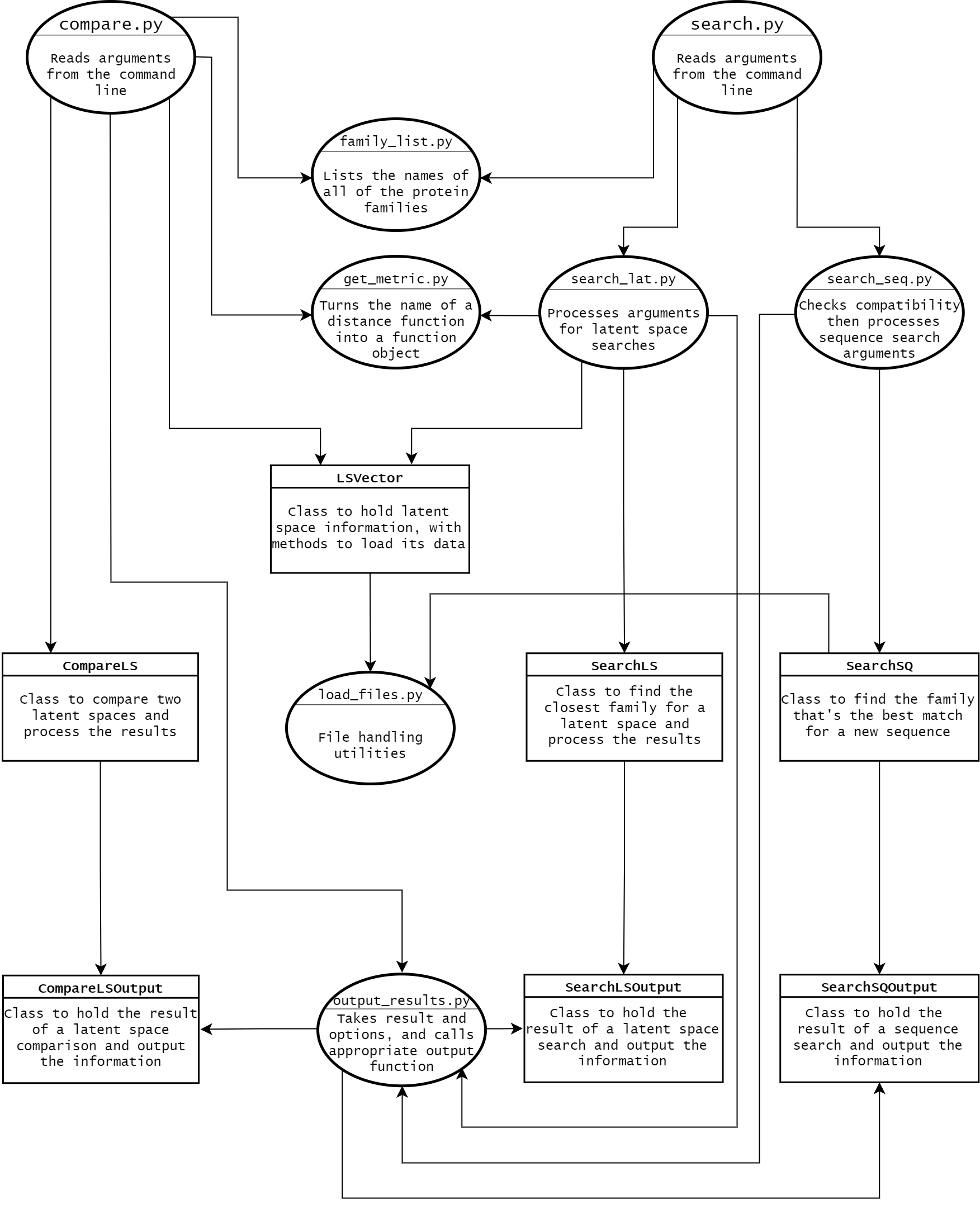
The file structure of the project as a whole was largely determined by the requirements for building Python
packages using setuptools.
All of the metadata needs to be in the base directory, with all of the code inside of folders for each
package.
Rather than listing each one in setup.cfg, we relied on setuptools's automatic package discovery, which
means each folder used as a package needs an __init__.py file inside of it.
We used a flat
layout, since there was only one root package.
CLI files
To the furthest extent possible we tried to isolate the CLI-specific code from the code that actually does
the work of the project, so it could be easily extended to support other input and output formats.
setup.cfg lists the two entry points as compare and search, which go
to the main functions in compare.py and search.py.
Inside of those files, argparse is used to process the input. To reduce any duplicated code, two
parent parsers are definied in __init__.py, and the other parsers inherit them.
One of them (dist_opt_parser) enables optional flags to specify which distance metric will be
used, and the other (output_opt_parser) allows users to choose between different forms of
output.
The current options allow output to be printed to stdout (default) or saved to a file, and files
can either include all of the human-readable text or just the values in csv format.
For files that already exist, users have the option to either overwrite that data or to append the new
information to the end of it.
search.py also uses subparsers, so the first argument after search selects which
parser to use.
One of them does latent space searches, and inherits both of the parent parsers, and the other handles
sequence searches and only inherits output_opt_parser.
A third subparser was added to print the full list of protein family names, but it doesn't accept any
arguments and functions more like a flag when used at the command line.
Each subparser goes to a different function after it's used, so there's no need to guess which one the users
used.
Both latent space searches and sequence searches can accept any number of filenames, which it appends to a
list stored in the argparse namespace.
compare.py only uses a single parser in the file, but it still inherits both of the parent
parsers.
Since users can give either a protein family name or a filename as input, it first tries treating it as a
protein family name, but if there isn't a family with that name it's treated as a filename.
It requires exactly two family / file names to be given as arguments, which it stores in a list in the
argparse namespace.
Since it requires two arguments, in order to list the known family names, users need to type list
names, which it then checks for before calling the comparison function.
Classes
The classes were designed to function independently of anything related to argparse, with a
series of helper functions bridging the gaps between the two sides.
LSVectors store the name of a latent space and its data, and there are functions to load the
data if it wasn't already given when the object was created.
CompareLS and SearchLS only take LSVectors and scipy distance
function objects as parameters, so it doesn't matter where the data inside of them came from.
The results returned from the comparisons and searches are in format-agnostic CompareLSOutput,
SearchLSOutput, and SearchSQOutput objects, which have methods for different ways
to output the results.
output_results.py uses the argparse arguments to determine which one should be
called, and since the method names are the same in all of the output classes it doesn't matter which one
it's operating on.